Vacuolar ATPase control of signaling and tumorigenesis
Commentary on Thomas Vaccari's paper published on Molecular oncology
September 2014
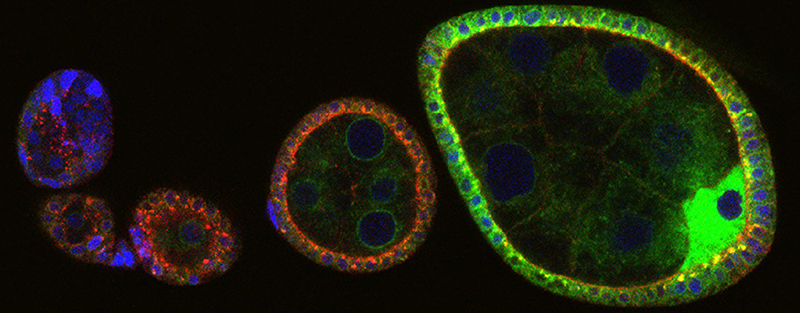
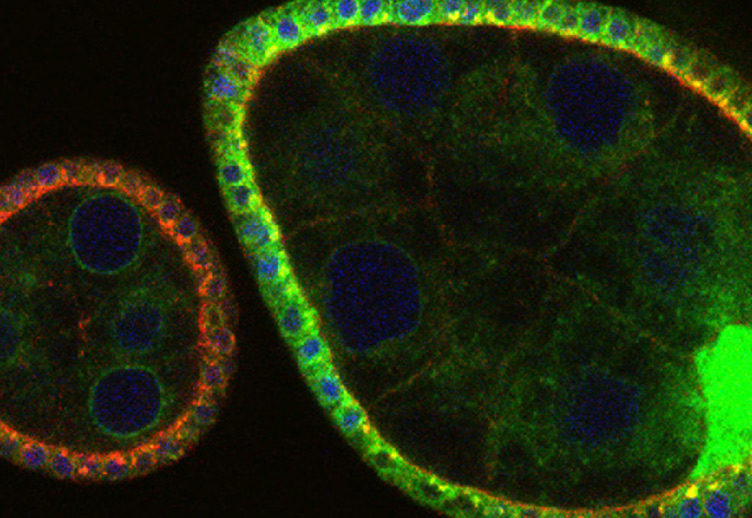
The study of most signal transduction pathways have progressed immensely in the last years in terms of identification of ligands, receptors, transducers, and transcriptional outputs. Yet, current knowledge of signaling circuitries is still vague in terms of how the parts interact with the cell infrastructure devoted to produce, deliver and degrade them, namely the membrane compartment. A case in point of such lack of cell biological insight is that of Notch signaling. This signaling system is simple, consisting of a transmembrane receptor that doubles as transcription factor, when liberated from membrane by a rather generic protease. The simplicity is only apparent considering that Notch signaling is used iteratively during organ development and homeostasis to instruct remarkably different cell fates such as survival, proliferation or differentiation, depending on the context. Part of context specificity is given by co-regulation with tissue-specific transcription factors. However, another part is likely to emerge from the way the receptor is handled by the protein trafficking machinery of the cell. The elegant work of Kobia and colleagues of Dr. Vaccari's group at IFOM now shows that inhibition of a key cellular enzyme, the vacuolar- H+ (V-) ATPase affects Notch signaling [1].
Notch signaling in prominently involved in growth regulation in metazoan tissues. Because of this, Notch is often upregulated in cancer and current efforts point to developing drugs that block its activation. Notch receptor endocytosis towards acidic compartments is a recently appreciated determinant of signaling activation. Vacuolar H(+) ATPase (V-ATPase) is responsible for acidification of endocytic organelles and mutants in V-ATPase subunit encoding genes in model organisms have been recently shown to display loss of Notch signaling. Here, we show that administration of BafilomycinA1 (BafA1), a highly specific V-ATPase inhibitor decreases Notch signaling during Drosophila and Zebrafish development, and in human cells in culture. In normal breast cells, we find that BafA1 treatment leads to accumulation of Notch in the endo-lysosomal system, and reduces its processing and signaling activity. In Notch-addicted breast cancer cells, BafA1 treatment reduces growth in cells expressing membrane tethered forms of Notch, while sparing cells expressing cytoplasmic forms. In contrast, we find that V-ATPase inhibition reduces growth of leukemia cells, without affecting Notch activatory cleavage. However, consistent with the emerging roles of V-ATPase in controlling multiple signaling pathways, in these cells Akt activation is reduced, as it is also the case in BafA1-treated breast cancer cells. Our data support V-ATPase inhibition as a novel therapeutic approach to counteract tumor growth via signaling pathways regulated at the endo-lysosomal level.
[PMID 24309677]
V-ATPase is the main energized proton pump in the cell. Its function is pleiotropic and central to many cellular processes such as organelle acidification and lysosomal function [2]. Despite more than 30 years of studies, V-ATPAse has only recently implicated in regulation of major signaling pathways [3,4]. In fact, the Vaccari lab had been one of the first groups who significantly contributed to this issue. Using Drosophila they showed that V-ATPase is required for activation of Notch signaling in the endo-lysosomal compartment [5]. Their and recent work by other investigators is of utmost importance: it is bringing into focus a complex network of regulation centered around trafficking and regulation of Notch degradation by the endo-lysosomal system, a recognized nexus for signaling regulation. Given the multiple roles that V-ATPase serves in cells, a number of outstanding interesting questions remain open. How is the enzyme regulated? How does it affect Notch signaling mechanistically? As far as I can tell from my fruitful interactions with Dr. Vaccari, his group is perfectly poised and highly competent to answer those questions experimentally.
Equally interesting is the relevance of work of Kobia and colleagues to cancer. Owing to the countless uses of Notch in shaping tissues, Notch disregulation is associated to multiple diseases. In particular, oncogenic Notch activation is a serious problem in T-cell acute lymphoblastic leukemia (T-ALL), and in certain breast and lung cancers. Kobia et al. demonstrated that pharmacologic block of V-ATPase with highly-specific compounds affects Notch signaling also in cancer cells with oncogenic Notch activity. Interestingly, while proliferation was reduced by V-ATPase inhibition in all cancer cell lines tested, Notch signaling was reduced only in cell lines derived from breast cancer and not from T-ALL. Cells in which Notch signaling was not reduced tended to be those in which Akt signaling, another major tumor-associated growth pathway, is perturbed. In these, V-ATPase inhibition mimicked the effects of Akt inhibitors. As a pharmacologist, I find inhibition of a pleiotropic enzyme a risky strategy to block oncogenes. It is indirect and one is bound to have plenty of side effects. This is why modern anticancer drugs are designed to be very specific. However, it is increasingly found that specific drugs induce resistance. Also, it is emerging that each tumor depends and is addicted to few signaling pathways acting redundantly. The work of Kobia et al. and work of my group on V-ATPase regulation of HER2 signaling [6] suggest that one could in fact hit more than one bird with one stone, perhaps limiting the ability of the tumor to "mutate away" from inhibition of a single signaling pathway.
A final interesting aspect of the recent work of Dr. Vaccari’s group is based on the fact that V-ATPase itself is found upregulated in a number of tumors. Despite previous work, whether upregulation is cause or consequence of tumorigenesis and how would V-ATPase would contribute to this is unresolved. In collaboration with a german group in Freiburg, the group of Thomas Vaccari has recently shown that overactivation of V-ATPase is sufficient for tissue transformation in Drosophila [7]. This, together with work of my group on V-ATPase and tumor cell migration [8], suggest that misregulation of V-ATPase might contribute to drive tissue transformation by altering multiple signaling pathways. Hopefully, we are going to learn much more of this in spring 2015 during the meeting "Vacuolar ATPase: A Novel Anti-Tumor Target" that I am co-organizing with Dr. Vaccari at IFOM.
Bibliography
- Kobia F, Duchi S, Deflorian G and Vaccari T (2014) Pharmacologic Inhibition Of Vacuolar H+ ATPase Reduces Physiologic And Oncogenic Notch Signaling. Molecular Oncology 8: 207-220.
- Nishi T and Forgac M (2002) The vacuolar (H+)-ATPases--nature's most versatile proton pumps. Nature Reviews Molecular Cell biology 3: 94-103.
- Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, et al. (2010) Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 327: 459-463.
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, et al. (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334: 678-683.
- Vaccari T, Duchi S, Cortese K, Tacchetti C and Bilder D (2010) The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development 137: 1825-1832.
- von Schwarzenberg K, Lajtos T, Simon L, Muller R, Vereb G, et al. (2014) V-ATPase inhibition overcomes trastuzumab resistance in breast cancer. Molecular Oncology 8: 9-19.
- Petzoldt AG, Gleixner EM, Fumagalli A, Vaccari T and Simons M (2013) Elevated expression of the V-ATPase C subunit triggers JNK-dependent cell invasion and overgrowth in a Drosophila epithelium. Disease Models & Mechanisms 6: 689-700.
- Wiedmann RM, von Schwarzenberg K, Palamidessi A, Schreiner L, Kubisch R, et al. (2012) The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res 72: 5976-5987