Cell therapy for muscular dystrophy: a step towards efficacy.
Commentary on Elisabetta Dejana's paper published on EMBO Molecular Medicine
February 2015
Background:
Duchenne Muscular Dystrophy (DMD) is severe and common, affecting one born male every 4,000. It is due to mutations in the dystrophin gene, the largest of our genome (larger than the whole genome of a bacterium), which was heroically cloned almost thirty years ago (Hoffman et al Cell 51,919,1987). It was then thought that, with the gene identified, the cure would have followed. It did not. The gene encodes for a large cytoskeletal protein that links the contractile apparatus to the membrane of the muscle fibre and adsorbs the mechanical stress of contraction. In its absence the membrane is damaged and the fibre dies. Initially, new fibres are formed by resident stem cells, termed “satellite cells” that share the same genetic defect and thus the same fate. If satellite cells are stem cells, they are amateur and not professionals like the hematopoietic stem cells, since muscles are made to last and do not turnover daily like blood. The sad evidence for this is observed in the biopsies of patients at late stages of the disease, where most muscles are gone, as are satellite cells, and have been replaced by scar and fat. At this stage no therapy will work. Patients progressively loose their motility and are confined to a wheelchair, followed by assisted ventilation and heart failure. Improved medical care has increased length and quality of life but a cure is still missing.
Muscular dystrophies are severe genetic diseases for which no efficacious therapies exist. Experimental clinical treatments include intra-arterial administration of vessel-associated stem cells, called mesoangioblasts (MABs). However, one of the limitations of this approach is the relatively low number of cells that engraft the diseased tissue, due, at least in part, to the sub-optimal efficiency of extravasation, whose mechanisms for MAB are unknown. Leukocytes emigrate into the inflamed tissues by crossing endothelial cell-to-cell junctions and junctional proteins direct and control leukocyte diapedesis. Here, we identify the endothelial junctional protein JAM-A as a key regulator of MAB extravasation. We show that JAM-A gene inactivation and JAM-A blocking antibodies strongly enhance MAB engraftment in dystrophic muscle. In the absence of JAM-A, the exchange factors EPAC-1 and 2 are down-regulated, which prevents the activation of the small GTPase Rap-1. As a consequence, junction tightening is reduced, allowing MAB diapedesis. Notably, pharmacological inhibition of Rap-1 increases MAB engraftment in dystrophic muscle, which results into a significant improvement of muscle function offering a novel strategy for stem cell-based therapies.
[PMID 24378569]
Cell therapy for muscular dystrophy.
In the last ten years the therapeutic landscape for DMD changed from a desert to a very busy field with many approaches entering clinical trials and some proceeding to Phase III (Mercuri & Muntoni Curr Op Neur 25,701,2013). Importantly, even if successful these therapies will be available only for subsets of patients (depending on the specific mutation) and many patients would not be eligible for any. This justifies the cell therapy approach that our group started many years. Skeletal muscle is stable and, if fixed, would require no further therapy. However it is the most abundant tissue of our body and therapy requires billions of cells. Moreover dystrophic muscle is affected by inflammation and fibrosis that make life hard for transplanted cells. Finally, since muscle fibres are multinucleated one healthy or “corrected” nucleus must work for all the others.
The previous work.
We tested cell transplantation in three mouse and one canine models of muscular dystrophy, using a cell type, termed “mesoangioblast” that we had identified in the blood vessel wall and characterized as a subset of pericytes, the cells that wrap the endothelium. The advantage of these cells, over resident satellite cells, that were subject of previous trials in the 90’, is their ability of crossing the vessel wall, when injected intra-arterially, which allow their even distribution in all the downstream muscles. However it appeared clear that in doing so, mesoangioblasts are less efficient than “professional” leukocytes, specifically endowed with all the molecular apparatus to get in and out the blood stream and reach the tissues where their action is needed. Nevertheless the results of the pre-clinical work were encouraging and led us to a “first in man” Phase I/IIa clinical trial with donor mesoangioblasts from an HLA-matched brother in five DMD patients. The trial (Cossu et al. in preparation) was safe but also showed limited efficacy. This was likely due to number of reasons, e.g. advanced age of patients and consequently a low level of engraftment, insufficient for a long-lasting significant clinical benefit. In a classic “from bench to bedside and back” approach, we are now dissecting each step of transplantation, aiming at increasing them all to finally reach efficacy.
The work we did.
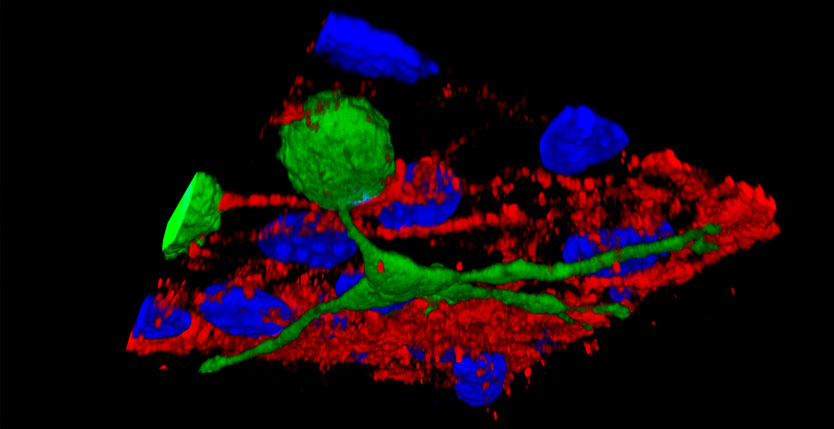
Among the steps to be improved, adhering to and crossing the endothelium are crucial because they determine what fraction of the injected cells actually gets to the muscle. JAM-A (Junctional Adhesion Molecule A) is an endothelial protein that tights the endothelium regulating cells transmigration. The group of Elisabetta Dejana at IFOM, long term collaborator, found that deleting this protein from the endothelium (or blocking it with a specific antibody) almost triples the amount of mesoangioblasts that cross the vessel wall both in vitro and in vivo (Giannotta et al EMM 6,239,2014). Notably, pharmacological inhibition of JAMA downstream effectors exerts the same effect thus suggesting possible drug-cells combined approach.
What next?
The natural follow up of the work on JAM-A would be clinical development of the inhibitor and/or the antibody, which however requires collaboration of a company. Alternatively, nanoparticles, charged with specific RNA inhibitors of JAM-A could be tested in mice: these particles have already been approved for clinical use and their clinical translation would thus faster, easier and cheaper.