Breaking up is hard to do: role of an endocytic adaptor protein in epithelial cell cohesion and breast tumor metastasis
Commentary on Giorgio Scita's paper published in Developmental Cell
May 2015
Epithelial cells are normally polarized and form extensive cell-cell contacts that allow for a barrier to be formed between apical and basolateral surfaces. Cadherin-based adherens junctions (AJs) are critical for epithelial cohesion, and alterations in cadherin expression or surface expression are hallmarks of epithelial-mesenchymal transition (EMT) and the acquisition of a motile phenotype. In metastatic epithelial cancers, the EMT program allows some tumor cells to breach local basement membrane barriers and reach blood or lymphatic vessels to disseminate to other tissues. A recent study by the Scita laboratory has implicated the F-BAR protein CIP4 (Cdc42-Interacting Protein-4) in mediating several key steps in breast cancer progression, including EMT, epithelial scattering, and breast cancer cell invasion (1). The authors also correlated high levels of CIP4 in human breast tumors with the highly metastatic HER2+ molecular subtype and risk of disease relapse.
The role of endocytic proteins and the molecular mechanisms underlying epithelial cell cohesion and tumor dissemination are not well understood. Here, we report that the endocytic F-BAR-containing CDC42-interacting protein 4 (CIP4) is required for ERBB2- and TGF-β1-induced cell scattering, breast cancer (BC) cell motility and invasion into 3D matrices, and conversion from ductal breast carcinoma in situ to invasive carcinoma in mouse xenograft models. CIP4 promotes the formation of an E-cadherin-CIP4-SRC complex that controls SRC activation, E-cadherin endocytosis, and localized phosphorylation of the myosin light chain kinase, thereby impinging on the actomyosin contractility required to generate tangential forces to break cell-cell junctions. CIP4 is upregulated in ERBB2-positive human BC, correlates with increased distant metastasis, and is an independent predictor of poor disease outcome in subsets of BC patients. Thus, it critically controls cell-cell cohesion and is required for the acquisition of an invasive phenotype in breast tumors.
[PMID 25203208]
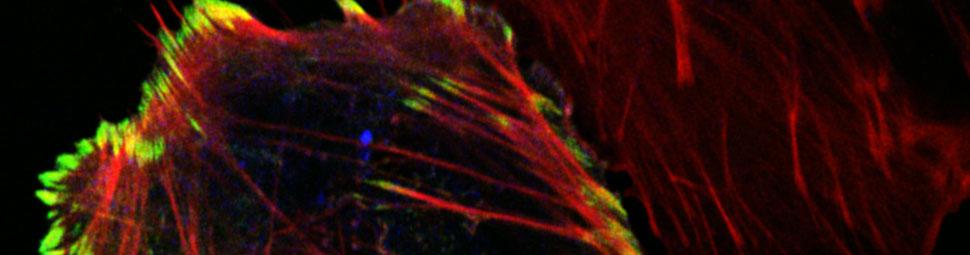
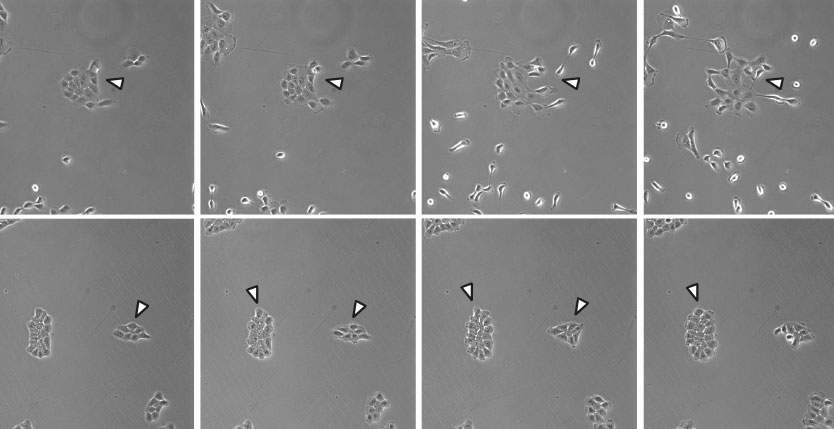
In normal breast epithelial cells, epidermal growth factor (EGF) induces cell scattering and invasion of extracellular matrix (ECM). However, silencing of CIP4 led to increased epithelial cell cohesion, and impaired cell scattering and invasion. This correlated with defects in actomyosin contractility in CIP4 knock-down (KD) cells, which was measured using an elegant tension sensor for E-Cadherin based on Forster resonance energy transfer. EGF-treated CIP4 KD cells displayed reduced tension across AJs compared to control cells, and reduced rates of E-cadherin internalization. In normal breast epithelial cells, EGF signaling induced CIP4 localization to AJs and binding and activation of Src protein-tyrosine kinase. Since Src promotes E-cadherin internalization and cell scattering, defects in junctional Src activation may explain the requirement for CIP4 in AJ dissolution. Together, these findings likely explain the increased epithelial cohesion and reduced cell scattering phenotypes observed in CIP4 KD cells.
To undergo EMT, epithelial cells exposed to TGF-β1 initiate a signaling pathway that targets junctional E-cadherin for rapid internalization, and cadherin switching marked by silencing of Cdh1 gene encoding E-cadherin, and upregulation of Cdh2 encoding N-cadherin. Consistent with some recent studies in other cell types, they show that TGF-β1 promotes CIP4 upregulation in breast epithelial cells, and CIP4 KD impairs the rate and extent of completing EMT in these cells. Mechanistically, there was no overt role for CIP4 in TGF-β1-induced activation of Smads, but Src activation was short-lived in CIP4 KD cells. This may contribute to non-canonical TGF-β1 signaling pathways that enhance expression of EMT drivers such as Snail1 and N-cadherin, which were both delayed or impaired in CIP4 KD cells.
To extend their study to human breast cancer, CIP4 expression was profiled in human breast tumor tissue microarrays. High levels of CIP4 was significantly associated with aggressive molecular subtypes (HER2+) and disease relapse. Interestingly, our group performed a similar study in an independent cohort of breast cancer patients, and found significantly increased risk of metastasis in patients with high CIP4 levels in their primary tumors (2). Since CIP4 was significantly associated with HER2+ tumors, Rolland and co-workers also tested the role of CIP4 in a model of ductal carcinoma in situ (DCIS)-to-invasive ductal carcinoma (IDC) conversion of MCF10A cells expressing an inducible ErbB2/HER2 allele. CIP4 silencing in this model impaired HER2-driven cell scattering and invasion, and limited DCIS-to-IDC conversion in mouse xenograft assays, based on loss of the myoepithelial marker alpha smooth muscle actin (1). Our study also tested the effects of CIP4 silencing in vivo, but focused instead on triple negative breast cancer (TNBC) models. We found that CIP4 silencing impairs TNBC metastasis to the lungs in mice (2).
Despite a relatively simple domain organization, it is interesting to note that CIP4 and related adaptor proteins play key roles in a growing list of diverse processes in normal cells and cancer cells. The study by Rolland and co-workers provides many fascinating insights into the localization and function of CIP4 in key steps in epithelial cell conversion to highly invasive and ultimately metastatic cancer cells. In future, it will be critical to fully elucidate the molecular mechanisms that could be best exploited to prevent metastasis and improve survival of cancer patients.
1. Rolland et al. (2014) The CDC42-interacting protein 4 controls epithelial cell cohesion and tumor dissemination. Dev Cell 30(5):553-68.
2. Cerqueira et al. (2015) CIP4 promotes metastasis in triple-negative breast cancer and is associated with poor patient prognosis. Oncotarget 6(11):9397-408.