Lymphomas: the backstage of the research that highlights their complexity and recommends the use of combined therapies
Commentary on Varano et al. "The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3β inhibition", Nature 2017, 546:302-6.
September 2017
Habent sua fata libelli - that's what came to my mind when I was asked to comment on the Varano et al. paper. In this case the special fatum is time scale and initial misfortune. The work started fifteen years ago, in 2002, in my laboratory at Harvard Medical School in Boston. A senior postdoc, Stefano Casola, and Simon Raffel, a German MD student, set out to explore whether what we had found for mature B cells in normal physiology also holds true for malignant B cells, namely that cellular survival depends on the expression of the B cell antigen receptor (BCR; Ref. 1). This was an important and therapeutically relevant question, and a clear answer came within a year or so, in the affirmative: In our mouse model of a MYC-driven B cell lymphoma complete cure was achieved when we inducibly deleted the BCR on the surface of the tumor cells in vivo! Fortunately we managed to stop publication of this spectacular result in a (very) high-ranking journal in the last minute: We had fallen into the trap of a technical artifact (2). And thus began the long and windy road towards the resolution of the problem. Still in Boston, Simon and Stefano showed that, surprisingly, in isolation BCR-negative lymphoma cells proliferated nearly as well as their BCR-positive counterparts; and that BCR-positive cells outcompeted BCR-negative cells in a variety of conditions. At that point Simon had to leave for his home university in Heidelberg, and in 2006 Stefano accepted a PI position at the IFOM in Milano. He built up his lab, hired students, wrote grants and embarked on numerous projects – but never gave up on studying the BCR dependency of B cell lymphomas, about which we continued to discuss. In the meantime the subject became popular and gained practical relevance, because B cell lymphoma therapies targeting the BCR signaling cascade were on the rise. They were not only in line with evidence from mouse models, but also directly supported, indeed suggested, by genetic experiments addressing the “Achilles heel” of human lymphoma cells in vitro (3).
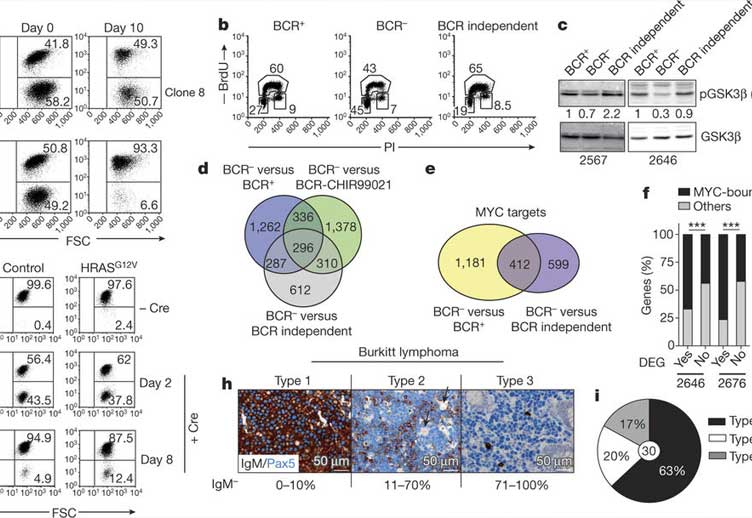
Similar to resting mature B cells, where the B-cell antigen receptor (BCR) controls cellular survival, surface BCR expression is conserved in most mature B-cell lymphomas. The identification of activating BCR mutations and the growth disadvantage upon BCR knockdown of cells of certain lymphoma entities has led to the view that BCR signalling is required for tumour cell survival. Consequently, the BCR signalling machinery has become an established target in the therapy of B-cell malignancies. Here we study the effects of BCR ablation on MYC-driven mouse B-cell lymphomas and compare them with observations in human Burkitt lymphoma. Whereas BCR ablation does not, per se, significantly affect lymphoma growth, BCR-negative (BCR-) tumour cells rapidly disappear in the presence of their BCR-expressing (BCR+) counterparts in vitro and in vivo. This requires neither cellular contact nor factors released by BCR+ tumour cells. Instead, BCR loss induces the rewiring of central carbon metabolism, increasing the sensitivity of receptor-less lymphoma cells to nutrient restriction. The BCR attenuates glycogen synthase kinase 3 beta (GSK3β) activity to support MYC-controlled gene expression. BCR- tumour cells exhibit increased GSK3β activity and are rescued from their competitive growth disadvantage by GSK3β inhibition. BCR- lymphoma variants that restore competitive fitness normalize GSK3β activity after constitutive activation of the MAPK pathway, commonly through Ras mutations. Similarly, in Burkitt lymphoma, activating RAS mutations may propagate immunoglobulin-crippled tumour cells, which usually represent a minority of the tumour bulk. Thus, while BCR expression enhances lymphoma cell fitness, BCR-targeted therapies may profit from combinations with drugs targeting BCR- tumour cells.
[PMID 28562582]
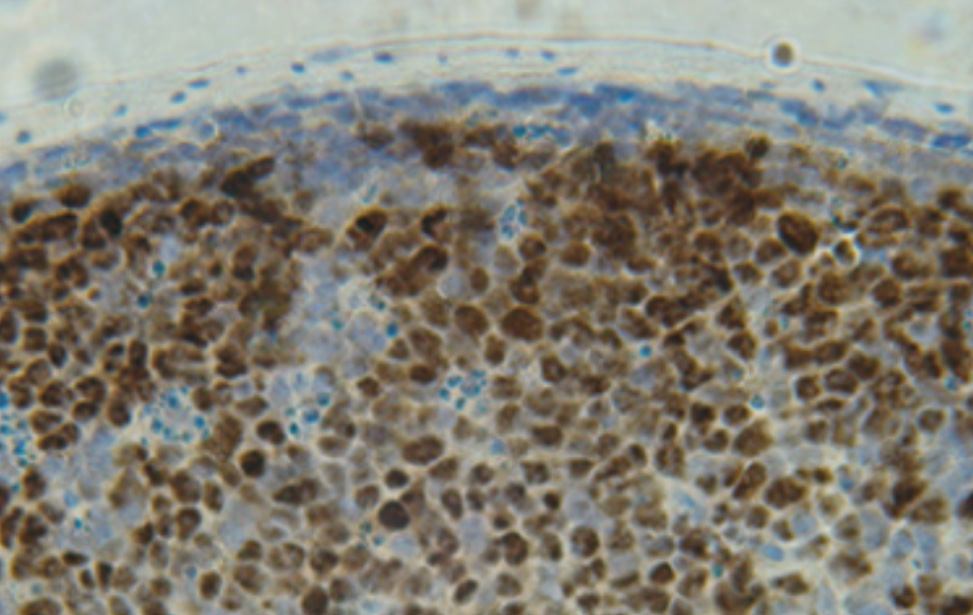
The painstaking experiments carried out in the Casola lab (Varano et al.) on the competition of BCR-positive and –negative MYC-driven lymphoma cells in mice add a new perspective to the multiple roles that BCR signaling presumably plays in transformed B cells. They suggest scenarios like that of human Burkitt lymphoma, where BCR expression impacts the fitness of the tumor cells in their competition with tumor cells that have lost the BCR. However, while the competitive advantage of the BCR-positive cells is indeed dramatic, variants of BCR-negative cells arise spontaneously and rapidly, often through RAS-mediated MAP kinase activation. The amazing efficiency of this process may be due to the surprising finding that BCR-mediated competitive fitness seems to critically depend on a single signaling pathway, namely PI3K/AKT dependent GSK3b inactivation, promoting MYC activity. Thus, therapeutic strategies targeting the BCR signaling cascade in B cell lymphomas may profit from complementary drugs targeting BCR loss variants. Note in this context that B cells are always in danger of losing BCR expression, given the mechanism of somatic hypermutation operating in these cells on antibody variable region genes to increase antibody affinity during antibody responses.
Beyond these translational considerations, the work of Varano et al. exemplifies the fundamental role of cellular competition in multicellular organisms in general and in our immune system in particular. To which extent and how wild type cells outcompete or actively eliminate spontaneously arising unwanted mutants, and whether such processes mainly apply to cancer pathogenesis or also operate in normal physiology remains a vast area for future research. Indeed, in the case of the B cell system it is striking that the central pathway controlling BCR-dependent lymphoma cell competitive fitness, namely PI3K signaling, has also been shown to control the survival and proliferation of normal B cells at various developmental stages, including that of the mature resting B cell (4).
Acknowledgements: I am grateful to Christine Kocks for critical reading.
References
1. Lam K-P, Kühn R and Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 1997, 90:1073-83
2. Schmidt-Supprian M and Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol 2007, 8:665-8
3. Davis RE et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463:88-92
4. Srinivasan L et al. PI3 Kinase signals BCR-dependent mature B cell survival. Cell 2009, 139:573-86