p53-dependent apoptosis: novel insight into the basis underlying microcephaly
A commentary on Vincenzo Costanzo's paper published in Nature Communications.
September 2016
Autosomal recessive primary microcephaly (MCPH) and Seckel Syndrome (SS) are overlapping disorders characterised by a reduced head circumference. MCPH has no or mild growth delay; growth delay in SS can be severe. Many causal genes for microcephaly encode centrosomal proteins although DNA damage response (DDR) genes have also been described. CEP63 is the causal defect in one MCPH/SS family with marked microcephaly and mild growth delay 1. CEP63 co-localises with pericentrin (PCNT), a well-studied centrosome protein, and regulates the centrosomal localisation of CEP152, a conserved centrosome duplication factor 2. CEP152/63 form a ring like structure around the parental centriole, and CEP63 loss in patients causes centrosome loss.
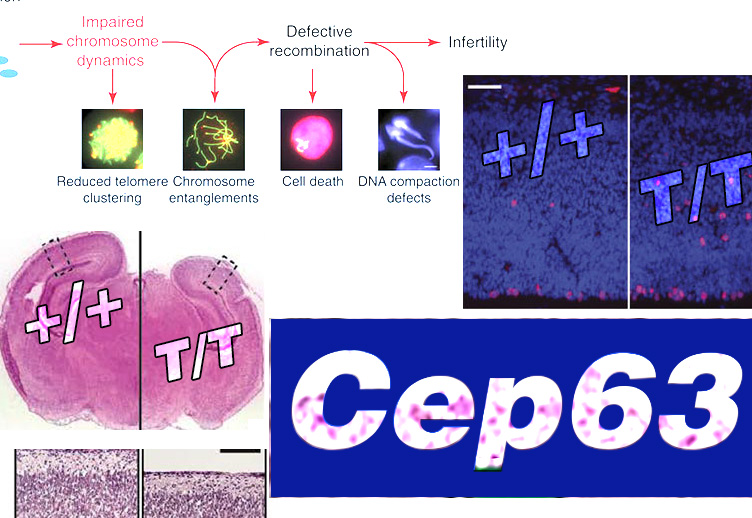
To investigate how CEP63 loss causes microcephaly, Costanzo, Stracker and colleagues examined neuronal development in mice with inactivated Cep63 3. Cep63 T/T, like CEP63 deficient patients, displayed growth delay and small head size. The mice showed abnormal Cep152 localisation in the embryonic neocortex and cells with monopolar spindles or abnormal spindle poles. Strikingly, in the embryonic neural stem cell region, the ventricular/subventricular zone (VZ/SVZ), the mitotic cell number was modestly increased with mitotic cells being frequently mislocalised. Enhanced apoptosis was observed throughout the neocortex. p53 is a DDR protein that regulates apoptosis. Strikingly, apoptosis was suppressed in p53-/-Cep63T/T mice and normal head size completely restored, although aberrant mitotic cell localisation remained. Thus, a striking finding is p53-dependent apoptotic activation arising from centrosome/mitotic abnormalities.
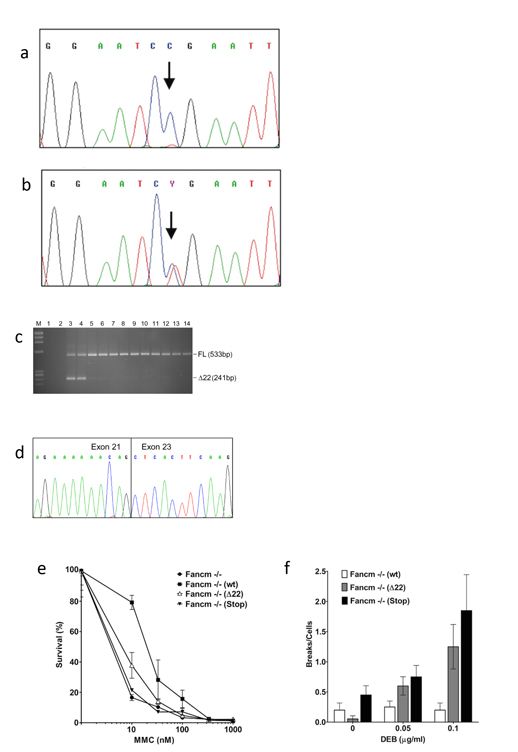
CEP63 is a centrosomal protein that facilitates centriole duplication and is regulated by the DNA damage response. Mutations in CEP63 cause Seckel syndrome, a human disease characterized by microcephaly and dwarfism. Here we demonstrate that Cep63-deficient mice recapitulate Seckel syndrome pathology. The attrition of neural progenitor cells involves p53-dependent cell death, and brain size is rescued by the deletion of p53. Cell death is not the result of an aberrant DNA damage response but is triggered by centrosome-based mitotic errors. In addition, Cep63 loss severely impairs meiotic recombination, leading to profound male infertility. Cep63-deficient spermatocytes display numerical and structural centrosome aberrations, chromosome entanglements and defective telomere clustering, suggesting that a reduction in centrosome-mediated chromosome movements underlies recombination failure. Our results provide novel insight into the molecular pathology of microcephaly and establish a role for the centrosome in meiotic recombination.
[PMID 26158450]
The embryonic VZ/SVZ cells proliferate rapidly from E11.5 to E16.5, initially via symmetric division to generate two daughter stem cells; subsequently, a switch to asymmetric division occurs producing a daughter destined to become a neuron. Premature switching to asymmetric division will diminish stem cell accrual and has been proposed as a mechanism underlying reduced brain size 4. Centrosome dysfunction has been proposed to promote premature switching. However, centrosome loss remains in p53-/-Cep63T/T mice suggesting that this cannot directly confer microcephaly. Cilia emanate from centrioles, the centrosome basal body and centrosome dysfunction impairs cilia signalling, providing a further possible causal mechanism. However, this defect will also remain in p53-/-Cep63T/T mice.
Elevated apoptosis correlates with microcephaly in other situations, such as radiation exposure or mouse models defective in the major DNA double strand break (DSB) repair pathway. LigIV-/- mice (DSB repair deficient) are embryonic lethal due to extensive neuronal apoptosis, which is rescued by p53 loss 5. Such apoptosis is DSB driven and ATM (a DDR kinase)-dependent; aberrant replication can activates ATR-dependent apoptosis. Yet DNA damage was not detected in Cep63T/T mice. Previous studies have observed p53 activation following centrosome loss. Importantly, two recent studies have provided mechanistic insight into a p53-dependent centrosome surveillance pathway, which is activated by centrosome loss or extended mitotic duration without detectable DNA damage 6, 7. p53 activation prevented cell cycle progression in the system examined.
So why does apoptotic induction preferentially confer microcephaly. Recent studies examining the haematopoietic system suggest that proliferating progenitors are sensitive to apoptosis whilst quiescent stem cells are resistant 8. Adult neural SVZ stem/progenitor cells appear to behave similarly 9. Rapid proliferation in the embryonic VZ/SVZ generates a large number of apoptotic-sensitive proliferating progenitor cells. Indeed, apoptosis in the VZ/SVZ of mice hypomorphic for LigIV is greater than in most other tissues 10. Thus, the embryonic brain may be exquisitely sensitive to apoptosis because it has an abundance of sensitive cells. Since replication ceases by E16.5, further progenitor replenishment may be precluded.
Cells from multiple SS patients display supernumerary centrosomes, which also likely prolongs mitosis 11. Thus, activation of apoptosis by centrosome abnormalities may be a common mechanism driving microcephaly in patients. However, abnormal mitoses persist in p53-/-Cep63T/T mice, potentially causing neuronal dysfunction despite normal head size. Patients lacking ATM do not show microcephaly but rather progressive ataxia, and it is tempting to speculate that the persistence of damaged neurons could be a contributing factor.
In summary, this significant study demonstrates that activation of p53-dependent apoptosis due to Cep63 loss confers microcephaly. This could represent a common mechanism for microcephaly since neuronal development produces many apoptotic-sensitive progenitor cells. Understanding the basis underlying microcephaly is currently important to evaluate the impact of the Zika virus.
References
1. Sir, J.H. et al. A primary microcephaly protein complex forms a ring around parental centrioles. Nature genetics 43, 1147-1153 (2011).
2. Brown, N.J., Marjanovic, M., Luders, J., Stracker, T.H. & Costanzo, V. Cep63 and cep152 cooperate to ensure centriole duplication. PloS one 8, e69986 (2013).
3. Marjanovic, M. et al. CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nature communications 6, 7676 (2015).
4. Thornton, G.K. & Woods, C.G. Primary microcephaly: do all roads lead to Rome? Trends in genetics : TIG 25, 501-510 (2009).
5. Frank, K.M. et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Molecular cell 5, 993-1002 (2000).
6. Lambrus, B.G. et al. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitotis. Journal of Cell Biology 214, 143-153 (2016).
7. Meitinger, F. et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. Journal of Cell Biology 214, 155-166 (2016).
8. Insinga, A. et al. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proceedings of the National Academy of Sciences of the United States of America 110, 3931-3936 (2013).
9. Daynac, M. et al. Quiescent neural stem cells exit dormancy upon alteration of GABAAR signaling following radiation damage. Stem cell research 11, 516-528 (2013).
10. Gatz, S.A. et al. Requirement for DNA ligase IV during embryonic neuronal development. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 10088-10100 (2011).
11. Alderton, G.K. et al. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Human molecular genetics 13, 3127-3138 (2004).